2.7 Download
All downloadable data provides users with a centralized access portal aimed at facilitating their access to research resources. We have integrated the following comprehensive datasets: (i) scATAC-seq data; (ii) Fine-mapping results; (iii) The trait–relevant score (TRS) of each single-cell generated by g-chromVAR and SCAVENGE methods; (iv) Results of gene and TF related analysis; (v) Gene regulation annotation data.
2.7.1 Download TRS data for each sample
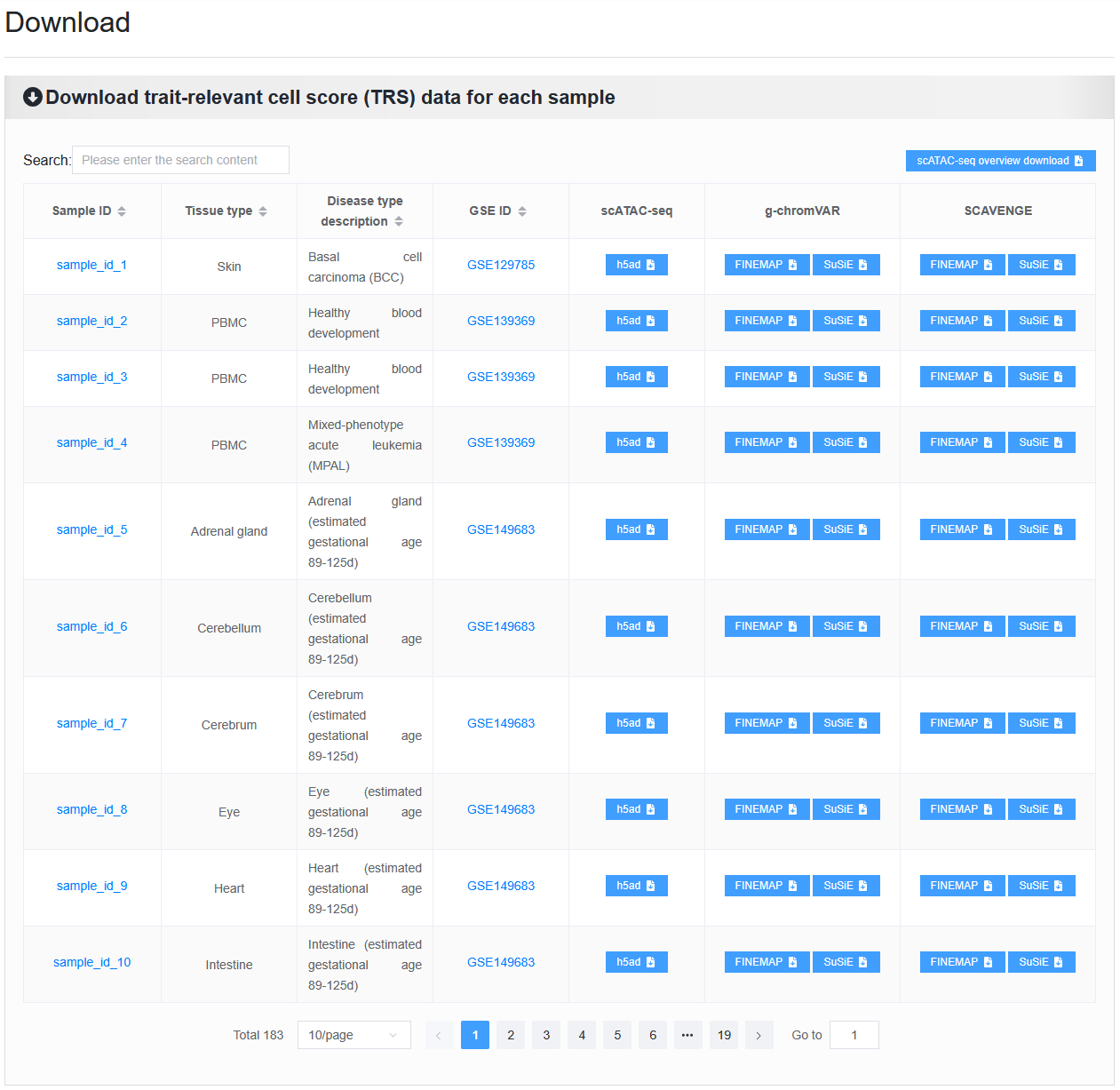
Below are the detailed download instructions.
2.7.1.1 Overview of scATAC-seq data: txt file
# |
Column name |
Description |
|---|---|---|
1 |
f_sample_id |
The unique identifier of the single-cell sample, used for database operations. |
2 |
f_gse_id |
GSE ID |
3 |
f_genome |
The reference genome of the single-cell sample. |
4 |
f_geo_id |
GEO ID |
5 |
f_label |
The unique identifier for the single-cell sample, used as the file name during data processing. |
6 |
f_pmid |
PMID |
7 |
f_species |
The species information of the single-cell sample. All data belongs to humans. |
8 |
f_tissue_type |
The tissue type of the single-cell sample. |
9 |
f_sequencing_type |
The sequencing type of the single-cell sample. |
10 |
f_health_type |
The health type of the single-cell sample. |
11 |
f_health_type_description |
Detailed information on the health type of the single-cell sample. |
12 |
f_description |
Detailed information on the content of the single-cell sample. |
13 |
f_source |
The source name of the single-cell sample. |
14 |
f_source_url |
The link to the source of the single-cell sample. |
15 |
f_counts_layer |
The layer name of the counts matrix stored in the Seurat object of the single-cell sample. |
16 |
f_sample_exist |
The single-cell sample contains multiple sample information. |
17 |
f_cell_count |
The number of cells in the single-cell sample. |
18 |
f_cell_type_count |
The number of cell types in the single-cell sample. |
19 |
f_index |
The unique index identifier of the single-cell sample has no meaning and is only used for sorting. |
20 |
f_time |
An indicator variable for whether this single-cell sample contains cell annotation information for age/day/time. 1 indicates presence, 0 indicates absence. |
21 |
f_sex |
An indicator variable for whether this single-cell sample contains cell annotation information for sex. 1 indicates presence, 0 indicates absence. |
22 |
f_drug |
An indicator variable for whether this single-cell sample contains cell annotation information for drug resistance. 1 indicates presence, 0 indicates absence. |
Note
When downloading files, some browsers will directly open the txt file and need to save the file by right-click.
2.7.1.2 scATAC-seq data: H5AD file
Read the information of sample_id_1.
1>>> data
2AnnData object with n_obs × n_vars = 36721 × 414680
3 obs: 'n_fragment', 'frac_dup', 'frac_mito', 'tsse', 'doublet_probability', 'doublet_score', 'barcode', 'n_genes', 'n_counts', 'cell_type', 'UMAP1', 'UMAP2', 'barcodes'
4 var: 'count', 'selected', 'chr', 'start', 'end', 'n_cells'
5 uns: 'doublet_rate', 'macs3', 'params', 'project_name', 'project_version', 'reference_sequences', 'scrublet_sim_doublet_score', 'step'
6 obsm: 'fragment_paired'
7>>>
8>>>
9>>> data.var
10 count selected chr start end n_cells
11index
12chr1:237500-238000 316.0 True chr1 237500 238000 296
13chr1:238000-238500 316.0 True chr1 238000 238500 296
14chr1:540500-541000 222.0 True chr1 540500 541000 217
15chr1:541000-541500 222.0 True chr1 541000 541500 217
16chr1:713500-714000 10773.0 True chr1 713500 714000 10145
17... ... ... ... ... ... ...
18chrX:155232500-155233000 246.0 True chrX 155232500 155233000 225
19chrX:155233500-155234000 200.0 True chrX 155233500 155234000 186
20chrX:155234000-155234500 200.0 True chrX 155234000 155234500 186
21chrX:155260000-155260500 603.0 True chrX 155260000 155260500 563
22chrX:155260500-155261000 603.0 True chrX 155260500 155261000 563
23
24[414680 rows x 6 columns]
25>>>
26>>>
27>>> data.obs
28 n_fragment frac_dup frac_mito tsse doublet_probability doublet_score barcode n_genes n_counts cell_type UMAP1 UMAP2 barcodes
29index
30AAACGAAAGAACGACC-1 24764 0.613793 0.0 14.751286 0.102154 0.095522 AAACGAAAGAACGACC-1 46094 49528 Tumor 4 10.567199 -4.781785 AAACGAAAGAACGACC-1
31AAACGAAAGAATACTG-1 2506 0.389822 0.0 14.333112 0.185441 0.001557 AAACGAAAGAATACTG-1 4809 5012 Myeloid 1.443223 13.324852 AAACGAAAGAATACTG-1
32AAACGAAAGACACGGT-1 4923 0.478827 0.0 23.241852 0.124562 0.040230 AAACGAAAGACACGGT-1 9438 9846 Treg -1.004199 -7.261578 AAACGAAAGACACGGT-1
33AAACGAAAGACCCTAT-1 3674 0.443755 0.0 21.428571 0.172410 0.007480 AAACGAAAGACCCTAT-1 7059 7348 B -5.697628 13.187097 AAACGAAAGACCCTAT-1
34AAACGAAAGAGGTACC-1 7178 0.488674 0.0 20.920746 0.152831 0.018101 AAACGAAAGAGGTACC-1 13666 14356 CD8 TEx -5.956334 -3.010488 AAACGAAAGAGGTACC-1
35... ... ... ... ... ... ... ... ... ... ... ... ... ...
36TTTGTGTTCGAGGCTC-1 4853 0.432597 0.0 17.623604 0.179749 0.004054 TTTGTGTTCGAGGCTC-1 9306 9706 Treg 1.477226 -8.637981 TTTGTGTTCGAGGCTC-1
37TTTGTGTTCGGGTCCA-1 5016 0.492256 0.0 24.892704 0.174884 0.006297 TTTGTGTTCGGGTCCA-1 9551 10032 Treg 2.348910 -6.036977 TTTGTGTTCGGGTCCA-1
38TTTGTGTTCGTCCCAT-1 12915 0.498855 0.0 15.457507 0.122509 0.042428 TTTGTGTTCGTCCCAT-1 24172 25830 CD8 TEx -8.256992 -3.043979 TTTGTGTTCGTCCCAT-1
39TTTGTGTTCTCTTCCT-1 5429 0.461569 0.0 19.229330 0.173898 0.006765 TTTGTGTTCTCTTCCT-1 10422 10858 Treg 2.174267 -8.784227 TTTGTGTTCTCTTCCT-1
40TTTGTGTTCTGCCGAG-1 3275 0.425842 0.0 16.528926 0.151769 0.018755 TTTGTGTTCTGCCGAG-1 6310 6550 Naive CD8 T -0.882584 1.916430 TTTGTGTTCTGCCGAG-1
41
42[36721 rows x 13 columns]
2.7.1.3 The result data of method g-ChromVAR: H5AD file
Read the information of sample_id_1 + FINEMAP.
obs: Cellvar: Trait or diseaseX: Z-score
1>>> data
2AnnData object with n_obs × n_vars = 36721 × 15805
3 obs: 'f_sample_id', 'f_barcodes', 'f_cell_type', 'f_sample', 'f_umap_x', 'f_umap_y', 'f_tsse', 'f_index', 'f_cell_type_index'
4 var: 'f_trait_id', 'f_trait_code', 'f_source_genome', 'f_trait_abbr', 'f_trait', 'f_variant_count'
5>>>
6>>> data.var
7 f_trait_id f_trait_code f_source_genome f_trait_abbr f_trait f_variant_count
8f_trait_id
9trait_id_826 trait_id_826 CAUSALdb_Appendicitis_PE06234_672 hg19 Appendicitis_PE06234 Appendicitis 13
10trait_id_2146 trait_id_2146 CAUSALdb_COE_FG02496_3096 hg19 COE_FG02496 Cancer of esophagus 2
11trait_id_3466 trait_id_3466 CAUSALdb_EHKPCAORROACYBNITLY_FG00466_5927 hg19 EHKPCAORROACYBNITLY_FG00466 Ever had known person concerned about, or reco... 1
12trait_id_1156 trait_id_1156 CAUSALdb_BNT_F900340_4465 hg19 BNT_F900340 Benign neoplasm: Testis 1
13trait_id_1816 trait_id_1816 CAUSALdb_CI_FG00089_4526 hg19 CI_FG00089 Carrot intake 21
14... ... ... ... ... ... ...
15trait_id_15801 trait_id_15801 UKBB_Worrier_43 hg19 Worrier Worrier 5683
16trait_id_15802 trait_id_15802 UKBB_Worry_Too_Long_85 hg19 Worry_Too_Long Worry too long after embarrassment 3225
17trait_id_15803 trait_id_15803 UKBB_eBMD_6 hg19 eBMD Estimated heel bone mineral density 37155
18trait_id_15804 trait_id_15804 UKBB_eGFR_15 hg19 eGFR Estimated glomerular filtration rate (serum cr... 35955
19trait_id_15805 trait_id_15805 UKBB_eGFRcys_3 hg19 eGFRcys Estimated glomerular filtration rate (cystain C) 37319
20
21[15805 rows x 6 columns]
22>>>
23>>> data.obs
24 f_sample_id f_barcodes f_cell_type f_sample f_umap_x f_umap_y f_tsse f_index f_cell_type_index
25index
26AAACGAAAGAACGACC-1 sample_id_1 AAACGAAAGAACGACC-1 Tumor 4 GSE129785 10.567199 -4.781785 14.751286 1 0
27AAACGAAAGAATACTG-1 sample_id_1 AAACGAAAGAATACTG-1 Myeloid GSE129785 1.443223 13.324852 14.333112 2 0
28AAACGAAAGACACGGT-1 sample_id_1 AAACGAAAGACACGGT-1 Treg GSE129785 -1.004199 -7.261578 23.241852 3 0
29AAACGAAAGACCCTAT-1 sample_id_1 AAACGAAAGACCCTAT-1 B GSE129785 -5.697628 13.187097 21.428571 4 0
30AAACGAAAGAGGTACC-1 sample_id_1 AAACGAAAGAGGTACC-1 CD8 TEx GSE129785 -5.956334 -3.010488 20.920746 5 0
31... ... ... ... ... ... ... ... ... ...
32TTTGTGTTCGAGGCTC-1 sample_id_1 TTTGTGTTCGAGGCTC-1 Treg GSE129785 1.477226 -8.637981 17.623604 36717 4065
33TTTGTGTTCGGGTCCA-1 sample_id_1 TTTGTGTTCGGGTCCA-1 Treg GSE129785 2.348910 -6.036977 24.892704 36718 4066
34TTTGTGTTCGTCCCAT-1 sample_id_1 TTTGTGTTCGTCCCAT-1 CD8 TEx GSE129785 -8.256992 -3.043979 15.457507 36719 3897
35TTTGTGTTCTCTTCCT-1 sample_id_1 TTTGTGTTCTCTTCCT-1 Treg GSE129785 2.174267 -8.784227 19.229330 36720 4067
36TTTGTGTTCTGCCGAG-1 sample_id_1 TTTGTGTTCTGCCGAG-1 Naive CD8 T GSE129785 -0.882584 1.916430 16.528926 36721 2767
37
38[36721 rows x 9 columns]
39>>>
40>>> data.X.todense()
41matrix([[ 0. , 0. , 0. , ..., 1.34798235,
42 0.13897425, 0.46950752],
43 [ 0. , 0. , 0. , ..., -0.27093183,
44 -0.28416698, 0.2759976 ],
45 [ 0. , 0. , 0. , ..., -0.6249468 ,
46 0.11480793, -1.2071487 ],
47 ...,
48 [ 0. , 0. , 0. , ..., -0.40784247,
49 0.35490693, -0.85452906],
50 [ 0. , 0. , 0. , ..., 0.50343663,
51 0.07536454, 0.42840868],
52 [ 0. , 0. , 0. , ..., -0.82765052,
53 0.20382107, 0.89792407]])
2.7.1.4 The result data of method SCAVENGE: H5AD file
Read the information of sample_id_1 + FINEMAP.
obs: Cellvar: Trait or diseaseX: TRS
1>>> data
2AnnData object with n_obs × n_vars = 36721 × 15805
3 obs: 'f_sample_id', 'f_barcodes', 'f_cell_type', 'f_sample', 'f_umap_x', 'f_umap_y', 'f_tsse', 'f_index', 'f_cell_type_index'
4 var: 'f_trait_id', 'f_trait_code', 'f_source_genome', 'f_trait_abbr', 'f_trait', 'f_variant_count'
5>>>
6>>>
7>>> data.X.todense()
8matrix([[0. , 0. , 0. , ..., 0.11992209, 0.26094234,
9 0.35693139],
10 [0. , 0. , 0. , ..., 0.50589785, 2.59232072,
11 1.68724861],
12 [0. , 0. , 0. , ..., 0.10034563, 0.40161146,
13 0.31860852],
14 ...,
15 [0. , 0. , 0. , ..., 0.03006235, 0.37951727,
16 0.08840483],
17 [0. , 0. , 0. , ..., 0.09616686, 0.52534063,
18 0.47852776],
19 [0. , 0. , 0. , ..., 0.21577299, 0.47587153,
20 0.39203965]])
21>>>
2.7.2 Download fine-mapping result data for each sample
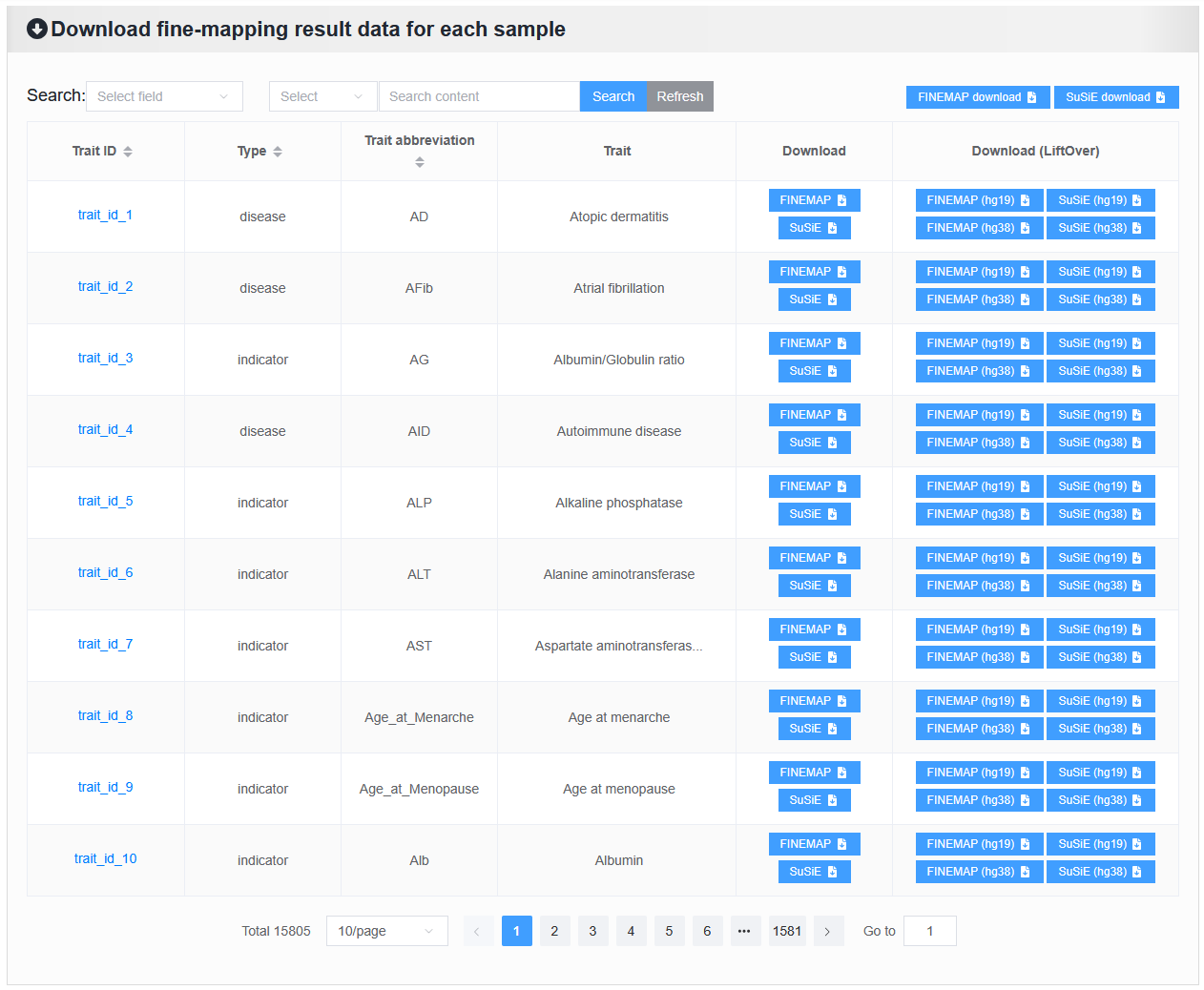
Below are the detailed download instructions.
2.7.2.1 Overview of fine-mapping result data: xlsx file
# |
Column name |
Description |
|---|---|---|
1 |
f_trait_id |
The unique identifier of the trait used for searching in the database. |
2 |
f_trait_index |
The unique identifier of the trait, used for sorting in the database, corresponds one-to-one with ‘f_trait_id’. |
3 |
f_trait_code |
The unique identifier of the trait, used as the file name for the file processing procedure. |
4 |
f_trait_abbr |
The abbreviation form of the trait. |
5 |
f_trait |
Detailed information for the trait. |
6 |
f_type |
The trait is classified as one of the types of “disease”, “drug”, “compound”, “health”, “subject”, “treatment”, “symptom”, “indicator” or “other”. |
7 |
f_icd10 |
ICD-10 |
8 |
f_category |
Major categories in ICD-10 |
9 |
f_sub_category |
Subcategories in ICD-10 |
10 |
f_three_category |
The third category in ICD-10 |
11 |
f_source_id |
Unique ID of the trait source cohort. |
12 |
f_source_name |
Name of the trait source cohort. |
13 |
f_source_genome |
Reference genome of trait source cohort. (Reference genome of the trait before LiftOver) |
14 |
f_variant_count |
The number of variant in the trait before LiftOver. |
15 |
f_variant_pp_sum |
The total PP value of variant in the trait before LiftOver. |
16 |
f_hg19_count |
The number of variant in the trait based on hg19 as a background reference genome. |
17 |
f_hg38_count |
The number of variant in the trait based on hg38 as a background reference genome. |
18 |
f_hg19_pp_sum |
The total PP value of variant in the trait based on hg19 as a background reference genome. |
19 |
f_hg38_pp_sum |
The total PP value of variant in the trait based on hg38 as a background reference genome. |
20 |
f_cohort |
The cohort for collecting the trait. |
21 |
f_author |
The author of the origin of the trait. |
22 |
f_mesh_id |
MESH ID |
23 |
f_mesh_term |
MESH TERM |
24 |
f_meta_id |
META ID |
25 |
f_popu |
Experimental population |
26 |
f_pmid |
PMID |
27 |
f_n_case |
Case size |
28 |
f_n_control |
Control size |
29 |
f_sample_size |
Sample size |
30 |
f_filter |
Each trait is retained, with a value of 1 for all. |
31 |
f_index |
The unique index identifier given in the same source cohort has no meaning and is only used to distinguish different traits in the same source cohort. |
32 |
f_url |
The link to download the source of each trait. |
2.7.2.2 Fine-mapping result data
txtfile (Downloadfield)
This file was formed through uniform processing after the original download.
# |
Column name |
Description |
|---|---|---|
1 |
trait_code |
unique identifier of the trait, used as the file name for the file processing procedure |
2 |
chr |
chromosome in the reference genome coordinate of the source cohort |
3 |
position |
position of variant in the reference genome coordinate of the source cohort |
4 |
variant |
unique variant identifier |
5 |
rsId |
rsID identifier |
6 |
allele1 |
reference allele in the reference genome coordinate of the source cohort |
7 |
allele2 |
alternative allele in the reference genome coordinate of the source cohort. (This allele is the effect allele.) |
8 |
maf |
allele frequency of the minor allele in cohort |
9 |
af |
allele frequency of allele2 (alt) |
10 |
beta |
marginal association effect size from linear mixed model/effect size GWAS |
11 |
se |
standard error on marginal association effect size from linear mixed model/standard error GWAS |
12 |
p_value |
p-value GWAS |
13 |
chisq |
test statistic for marginal association |
14 |
z_score |
original z-score |
15 |
pp |
posterior probability of association from fine-mapping (FINEMAP or SuSiE) |
16 |
beta_posterior |
posterior expectation of true effect size |
17 |
sd_posterior |
posterior standard deviation of true effect size |
18 |
trait_abbr |
abbreviation for the trait |
19 |
trait |
detailed information for the trait |
20 |
index |
Unique index identifiers based on trait or disease variants are meaningless and can be used to identify the uniqueness of variants. |
Note
When collecting fine-mapping result data, some data may not include all columns, and a small number of columns may have null values. Of course, the four columns of “chr”, “position”, “pp”, and “trait” are definitely included.
bedfile (Download (LiftOver)field)
scVMAP provides variant coordinates under different reference genomes.
# |
Column name |
Description |
|---|---|---|
1 |
None |
chromosome in hg19/hg38 coordinates |
2 |
None |
(start) position of variant in hg19/hg38 coordinates |
3 |
None |
(end) position of variant in hg19/hg38 coordinates |
4 |
None |
rsID identifier |
5 |
None |
posterior probability of association from fine-mapping (FINEMAP or SuSiE) |
6 |
None |
abbreviation for the trait |
7 |
None |
Unique index identifiers based on trait or disease variants are meaningless and can be used to identify the uniqueness of variants. |
Note
This format of data is suitable for performing overlay operations with enhancer data, etc.
Note
The download name is the same regardless of the method or reference genome selected, so please be aware of this.
2.7.3 Download other data
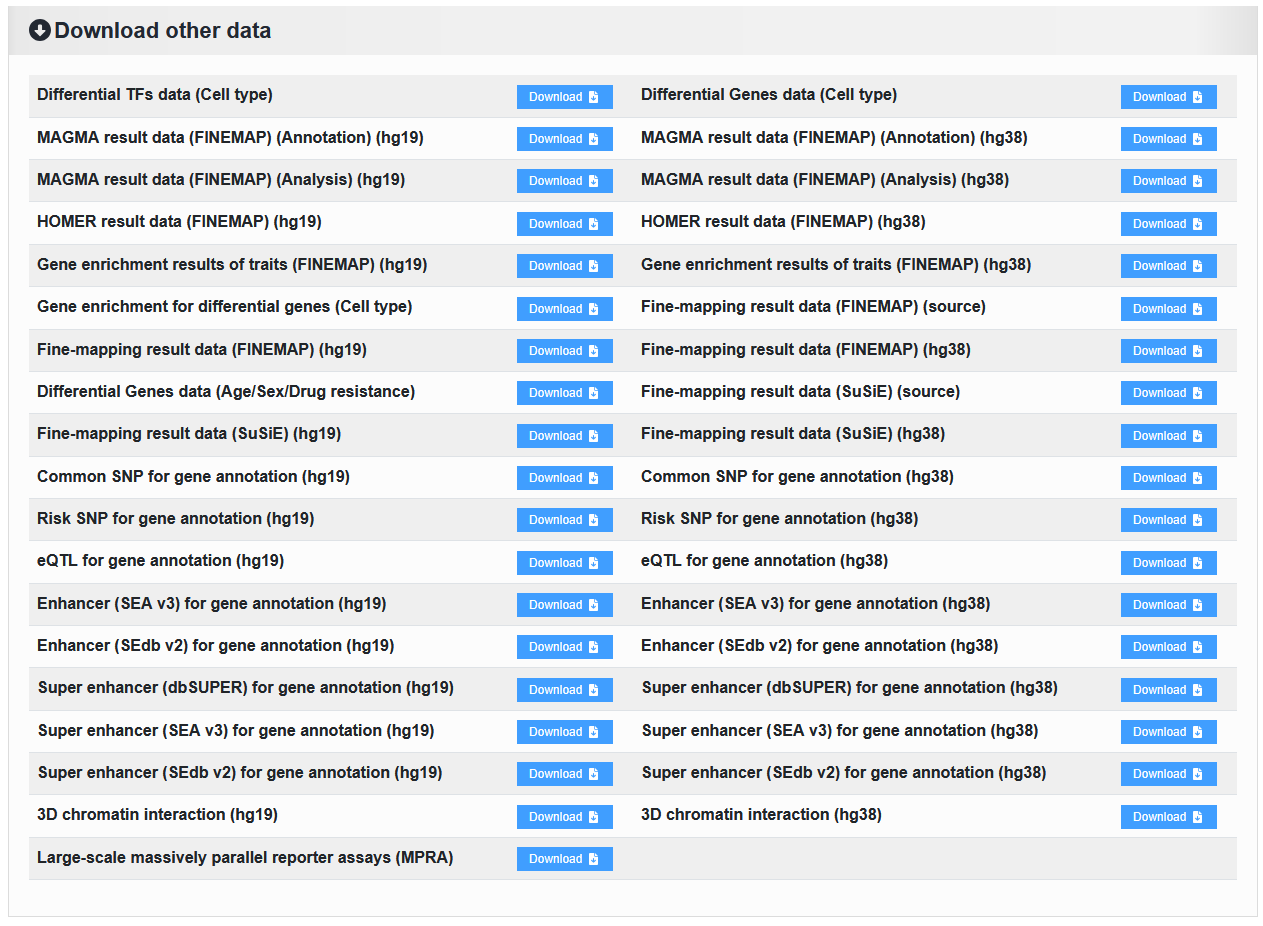
2.7.3.1 Fine-mapping result data: tar.gz file
Here is the complete download for Part 2.7.2 Download fine-mapping result data for each sample.
Fine-mapping result data (FINEMAP/SuSiE) (source):txtfile (Downloadfield)Repeat display once:
# |
Column name |
Description |
|---|---|---|
1 |
trait_code |
unique identifier of the trait, used as the file name for the file processing procedure |
2 |
chr |
chromosome in the reference genome coordinate of the source cohort |
3 |
position |
position of variant in the reference genome coordinate of the source cohort |
4 |
variant |
unique variant identifier |
5 |
rsId |
rsID identifier |
6 |
allele1 |
reference allele in the reference genome coordinate of the source cohort |
7 |
allele2 |
alternative allele in the reference genome coordinate of the source cohort. (This allele is the effect allele.) |
8 |
maf |
allele frequency of the minor allele in cohort |
9 |
af |
allele frequency of allele2 (alt) |
10 |
beta |
marginal association effect size from linear mixed model/effect size GWAS |
11 |
se |
standard error on marginal association effect size from linear mixed model/standard error GWAS |
12 |
p_value |
p-value GWAS |
13 |
chisq |
test statistic for marginal association |
14 |
z_score |
original z-score |
15 |
pp |
posterior probability of association from fine-mapping (FINEMAP) |
16 |
beta_posterior |
posterior expectation of true effect size |
17 |
sd_posterior |
posterior standard deviation of true effect size |
18 |
trait_abbr |
abbreviation for the trait |
19 |
trait |
detailed information for the trait |
20 |
index |
Unique index identifiers based on trait or disease variants are meaningless and can be used to identify the uniqueness of variants. |
Fine-mapping result data (FINEMAP/SuSiE) (hg19/hg38):bedfile (Download (LiftOver)field)Repeat display once:
# |
Column name |
Description |
|---|---|---|
1 |
None |
chromosome in hg19/hg38 coordinates |
2 |
None |
(start) position of variant in hg19/hg38 coordinates |
3 |
None |
(end) position of variant in hg19/hg38 coordinates |
4 |
None |
rsID identifier |
5 |
None |
posterior probability of association from fine-mapping (FINEMAP or SuSiE) |
6 |
None |
abbreviation for the trait |
7 |
None |
Unique index identifiers based on trait or disease variants are meaningless and can be used to identify the uniqueness of variants. |
2.7.3.2 Differential gene data: txt file
Differential Genes data (Cell type):tar.gzfile
This file contains differential gene data for all cell types of single-cell samples. Of course, it is after passing the threshold.
# |
Column name |
Description |
|---|---|---|
1 |
f_sample_id |
unique identifier of scATAC-seq sample |
2 |
f_cell_type |
cell type |
3 |
f_gene |
gene name |
4 |
f_score |
score |
5 |
f_adjusted_p_value |
adjusted p value |
6 |
f_log2_fold_change |
Log2(Fold change) |
7 |
f_p_value |
P-value |
Differential Genes data (Age/Sex/Drug resistance):txtfile
This file contains differential gene data for all cell types of single-cell samples. Of course, it is after passing the threshold.
# |
Column name |
Description |
|---|---|---|
1 |
f_sample_id |
unique identifier of scATAC-seq sample |
2 |
f_type_value |
Corresponds to the values under the f_type field. |
3 |
f_gene |
gene name |
4 |
f_score |
score |
5 |
f_adjusted_p_value |
adjusted p value |
6 |
f_log2_fold_change |
Log2(Fold change) |
7 |
f_p_value |
P-value |
7 |
f_type |
Age, gender, or drug resistance information. |
Note
You need to download the complete data without threshold filtering, and enter the details page of the sample to download the H5AD file.
Example: sample_id_1
1>>> data
2AnnData object with n_obs × n_vars = 33501 × 20
3 obs: 'n_cells'
4 var: 'cell_type', 'size'
5 uns: 'diff_genes'
6 layers: 'adjusted_p_value', 'log2_fold_change', 'p_value'
7>>>
8>>> data.var
9 cell_type size
10cell_type
11B B 404
12CD8 TEx CD8 TEx 3898
13Effector CD8 T Effector CD8 T 1153
14Endothelial Endothelial 562
15Fibroblasts Fibroblasts 1325
16Memory CD8 T Memory CD8 T 4965
17Myeloid Myeloid 732
18NK1 NK1 418
19NK2 NK2 1207
20Naive CD4 T Naive CD4 T 4059
21Naive CD8 T Naive CD8 T 2768
22Plasma B Plasma B 335
23Tfh Tfh 4138
24Th1 Th1 338
25Th17 Th17 1842
26Treg Treg 4068
27Tumor 1 Tumor 1 757
28Tumor 2 Tumor 2 875
29Tumor 3 Tumor 3 1687
30Tumor 4 Tumor 4 1190
31>>>
32>>> data.obs
33 n_cells
34AP006222.2 296
35ENSG00000286448 296
36ENSG00000230021 14992
37ENSG00000228327 10389
38LINC01409 10389
39... ...
40TMLHE 4231
41SPRY3 5205
42VAMP7 7748
43IL9R 5738
44ENSG00000270726 395
45
46[33501 rows x 1 columns]
47>>>
48>>> data.X
49array([[-16.08996773, 16.2977314 , -3.94544339, ..., 22.60018349,
50 65.58148956, 41.31241226],
51 [ -9.23847771, 38.57592773, -28.23983192, ..., -8.53127384,
52 16.334095 , 46.58874512],
53 [ -9.22247505, 38.53868484, -28.31791878, ..., -8.08869743,
54 16.5304184 , 46.68078613],
55 ...,
56 [ -0.73027158, 34.58570862, 42.81091309, ..., -33.24862289,
57 -56.29743958, -51.4512825 ],
58 [ 12.86117554, -13.21335506, -1.77498877, ..., -29.03244019,
59 -39.19504929, -43.00321579],
60 [-16.56791496, -32.8029213 , 2.89613366, ..., 38.49712753,
61 32.102005 , -17.40989685]])
62>>>
2.7.3.3 Differential TF data: txt file
This file contains differential TF data for all cell types of single-cell samples. Of course, it is after passing the threshold.
# |
Column name |
Description |
|---|---|---|
1 |
f_sample_id |
unique identifier of scATAC-seq sample |
2 |
f_cell_type |
cell type |
3 |
f_tf |
transcription factor name |
4 |
f_tf_id |
unique identifier of transcription factor |
5 |
f_p_value |
P-value |
6 |
f_adjusted_p_value |
adjusted p value |
7 |
f_log2_fold_change |
Log2(Fold change) |
Note
You need to download the complete data without threshold filtering, and enter the details page of the sample to download the H5AD file.
Example: sample_id_1
1>>> data
2AnnData object with n_obs × n_vars = 1165 × 20
3 obs: 'id', 'name'
4 var: 'cell_type', 'size'
5 layers: 'adjusted_p_value', 'log2_fold_change'
6>>>
7>>> data.obs
8 id name
9index
10AC023509.3+M02872_2.00 AC023509.3+M02872_2.00 AC023509.3
11AC138696.1+M04597_2.00 AC138696.1+M04597_2.00 AC138696.1
12AHR+M09817_2.00 AHR+M09817_2.00 AHR
13AIRE+M09375_2.00 AIRE+M09375_2.00 AIRE
14ALX1+M05327_2.00 ALX1+M05327_2.00 ALX1
15... ... ...
16ZSCAN4+M02919_2.00 ZSCAN4+M02919_2.00 ZSCAN4
17ZSCAN5+M04460_2.00 ZSCAN5+M04460_2.00 ZSCAN5
18ZSCAN5C+M08390_2.00 ZSCAN5C+M08390_2.00 ZSCAN5C
19ZSCAN9+M04466_2.00 ZSCAN9+M04466_2.00 ZSCAN9
20ZZZ3+M01272_2.00 ZZZ3+M01272_2.00 ZZZ3
21
22[1165 rows x 2 columns]
23>>>
24>>> data.X
25array([[1.01662951e-01, 1.74660328e-01, 2.50931395e-01, ...,
26 6.34538848e-02, 7.25013930e-02, 5.10951651e-05],
27 [2.07562180e-01, 1.93983057e-01, 2.10357488e-01, ...,
28 3.01950908e-01, 3.46950746e-01, 8.56932171e-02],
29 [2.40413032e-01, 9.76634287e-02, 6.66147596e-01, ...,
30 2.68301581e-01, 1.75328527e-02, 1.26211337e-03],
31 ...,
32 [4.38363454e-01, 1.43397437e-01, 4.24778841e-01, ...,
33 7.15759727e-03, 5.41759614e-02, 9.35845828e-12],
34 [4.86767592e-01, 1.47841135e-01, 5.32381338e-01, ...,
35 2.74014131e-01, 1.13489445e-05, 6.38005942e-11],
36 [1.61418404e-01, 3.23724955e-01, 4.50586827e-02, ...,
37 2.66768124e-01, 7.84328678e-02, 4.08885306e-07]])
38>>>
2.7.3.4 MAGMA result data: tar.gz file
The result data of enriched genes for traits or diseases through MAGMA.
MAGMA result data (Annotation) (hg19/hg38):AnnotationMAGMA result data (Analysis) (hg19/hg38):Gene analysis -raw data
2.7.3.4.1 Annotation: txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
trait_id |
unique identifier of trait or disease |
2 |
gene_id |
unique identifier of gene |
3 |
gene |
gene name |
4 |
rsId |
rsID identifier |
Note
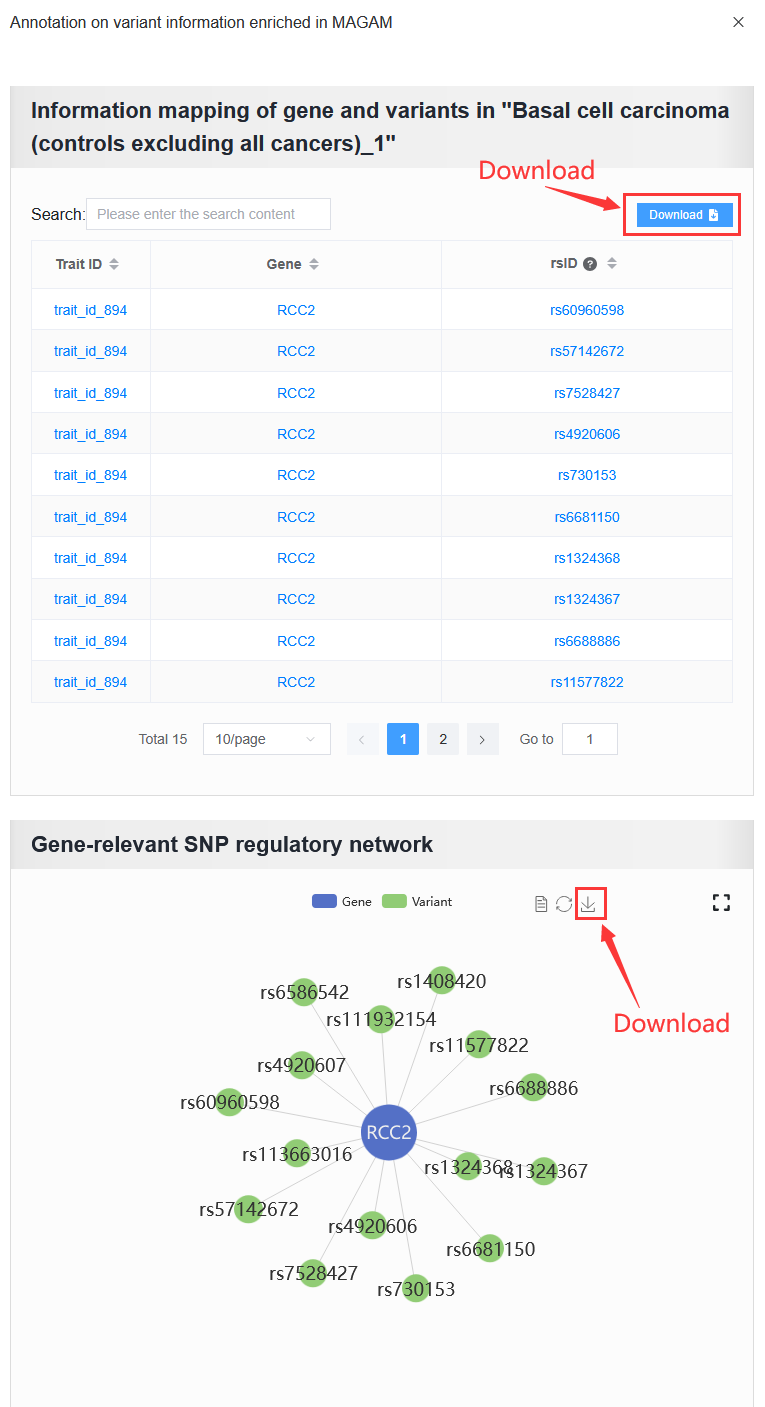
The user needs to obtain the genes.annot file after MAGMA runs and needs to enter the details page to obtain it.
Example: trait_id_894
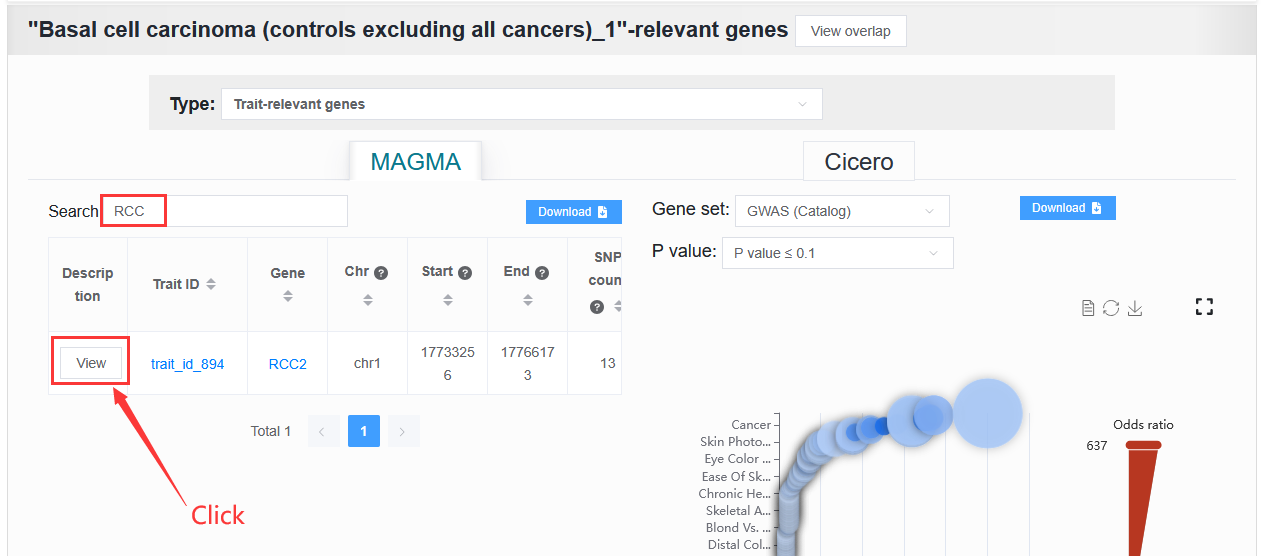
Click View
2.7.3.4.1 Gene analysis -raw data: txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
trait_id |
unique identifier of trait or disease |
2 |
gene_id |
unique identifier of gene |
3 |
gene |
gene name |
4 |
chr |
chromosome code |
5 |
start |
starting boundary of gene annotation on chromosomes |
6 |
end |
ending boundary of gene annotation on chromosomes |
7 |
n_snps |
The number of SNPs not annotated to this gene based on previous SNP QC exclusion. |
8 |
z_score |
z-value |
9 |
p_value |
p-value |
Note
The user needs to obtain the genes.out file after MAGMA runs and needs to enter the details page to obtain it.
Example: trait_id_894
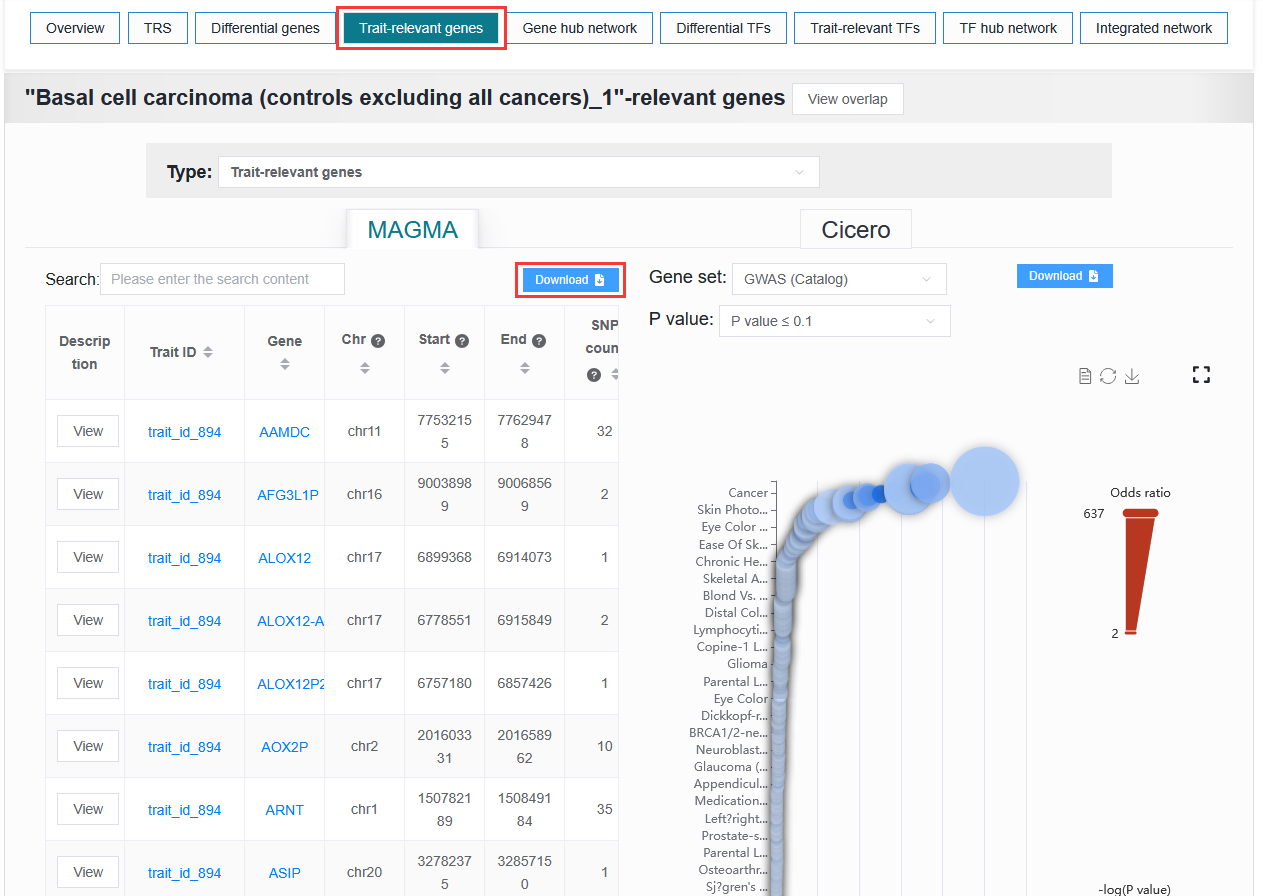
2.7.3.5 HOMER result data: tar.gz file
HOMER result data (hg19/hg38):txtfile (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
f_trait_id |
unique identifier of trait or disease |
2 |
f_motif_name |
unique identifier of gene |
3 |
f_tf |
TF name |
4 |
f_consensus |
consensus |
5 |
f_p_value |
p-value |
6 |
f_q_value |
q-value |
Note
Users need to download complete data without threshold filtering and enter the details page to download the file.
Example: trait_id_894
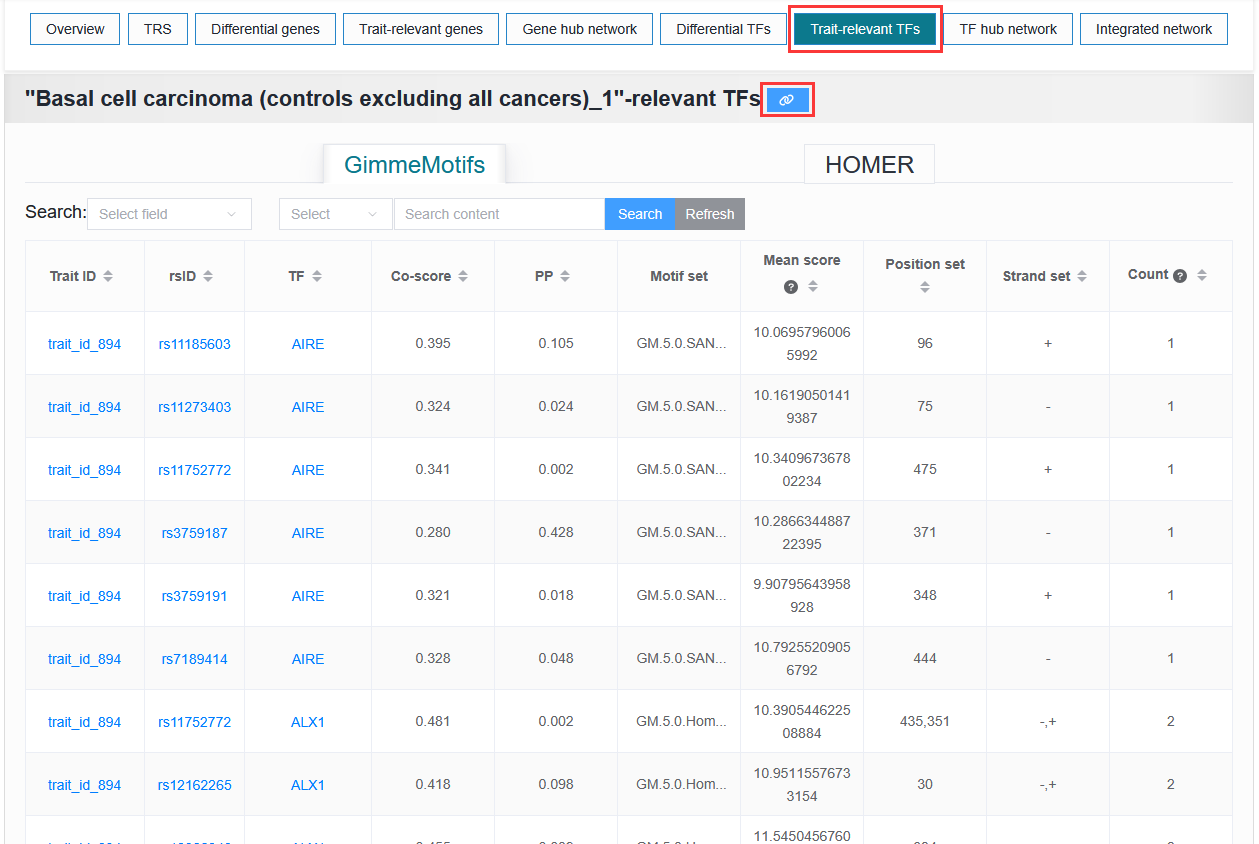
Click on the link symbol button.
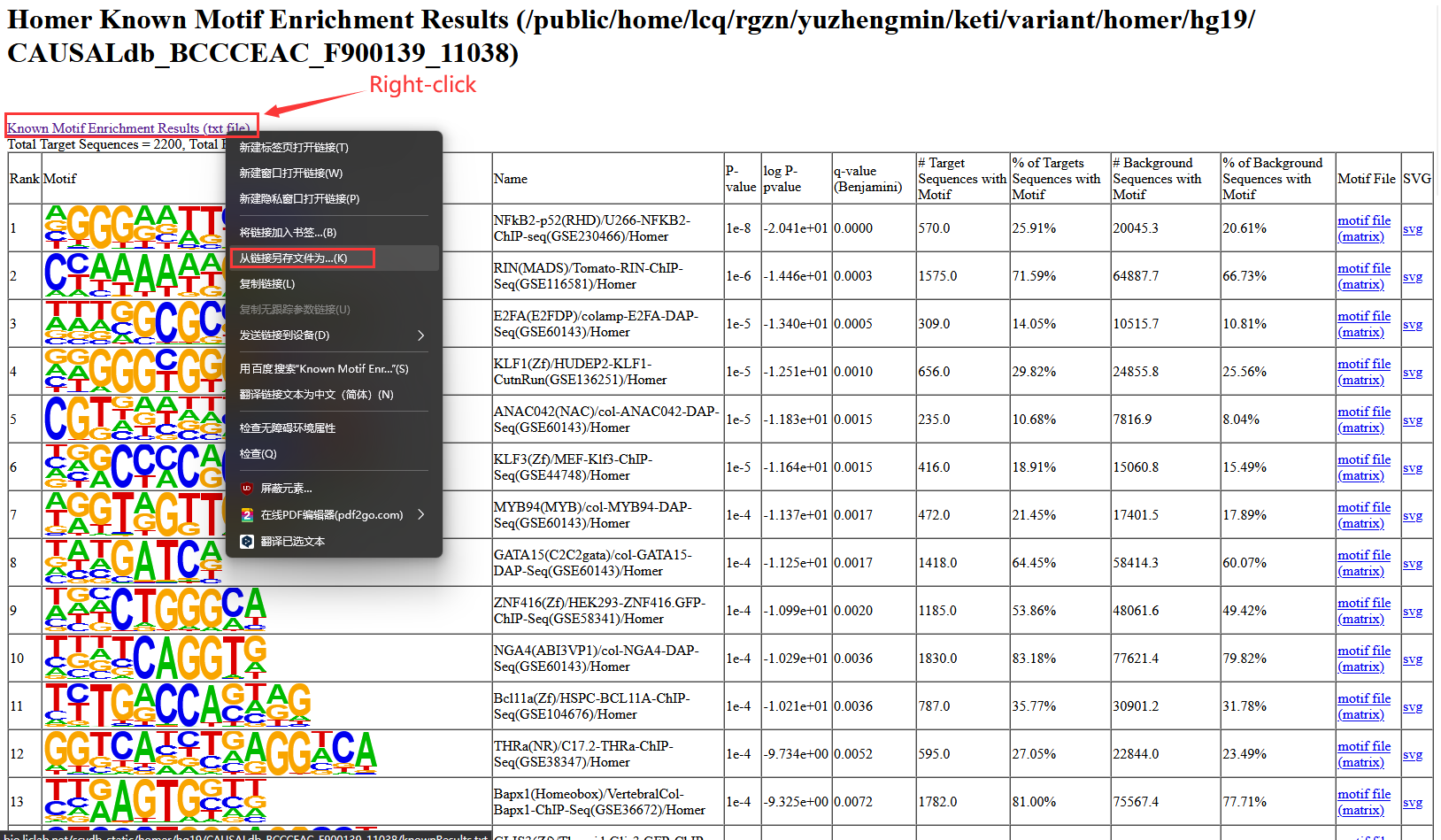
2.7.3.6 Gene enrichment analysis results: tar.gz file
Gene enrichment for differential genes:txtfile (After decompression)Gene enrichment results of traits (hg19/hg38):txtfile (After decompression)
2.7.3.6.1 Gene enrichment for differential genes
File name: {Sample ID}_gene_enrichment_data.txt
# |
Column name |
Description |
|---|---|---|
1 |
f_gene_set |
Gene set (GO_Biological_Process_2023, GO_Cellular_Component_2023, GO_Molecular_Function_2023 and GWAS_Catalog_2023) |
2 |
f_term |
gene enrichment term |
3 |
f_overlap |
percentage of gene set overlap |
4 |
f_p_value |
p-value |
5 |
f_adjusted_p_value |
adjusted p-value |
6 |
f_odds_ratio |
odds ratio |
7 |
f_combined_score |
combined score |
8 |
f_gene |
overlap genes |
9 |
f_count |
count of overlapping genes |
10 |
f_cell_type |
cell type |
2.7.3.6.2 Gene enrichment results of traits (hg19/hg38)
File name: {Trait label}_gene_enrichment_trait_data.txt
# |
Column name |
Description |
|---|---|---|
1 |
trait_id |
unique identifier of trait or disease |
2 |
Gene_set |
Gene set (GO_Biological_Process_2023, GO_Cellular_Component_2023, GO_Molecular_Function_2023 and GWAS_Catalog_2023) |
3 |
Term |
gene enrichment term |
4 |
Overlap |
percentage of gene set overlap |
5 |
P-value |
p-value |
6 |
Adjusted P-value |
adjusted p-value |
7 |
Old P-value |
old p-value |
8 |
Old Adjusted P-value |
old adjusted p-value |
9 |
Odds Ratio |
odds ratio |
10 |
Combined Score |
combined score |
11 |
Genes |
overlap genes |
Note
A very small number of traits or diseases contain too few fine-mapped variants, resulting in a lack of gene enrichment results.
2.7.3.7 Gene regulation/V2G annotation data:
scVMAP provides gene regulation annotation data for five types of epigenome data.
2.7.3.7.1 Common SNP: txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
chr |
chromosome |
2 |
position |
position |
3 |
rsId |
rsID identifier |
4 |
ref |
reference allele in the reference genome coordinate of the source cohort |
5 |
alt |
alternative allele in the reference genome coordinate of the source cohort. (This allele is the effect allele.) |
1$ head dbsnp_common_snp_hg38.txt
2chr position rsId ref alt
3chr1 10177 rs367896724 A AC
4chr1 10352 rs555500075 T TA
5chr1 10616 rs376342519 CCGCCGTTGCAAAGGCGCGCCG C
6chr1 11012 rs544419019 C G
7chr1 11063 rs561109771 T G
8chr1 13110 rs540538026 G A
9chr1 13116 rs62635286 T G
10chr1 13118 rs62028691 A G
11chr1 13273 rs531730856 G C
2.7.3.7.2 eQTL: txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
chr |
chromosome |
2 |
position |
position |
3 |
ref |
reference allele in the reference genome coordinate of the source cohort |
4 |
alt |
alternative allele in the reference genome coordinate of the source cohort. (This allele is the effect allele.) |
5 |
gene_name |
gene name |
6 |
tss_distance |
The distance between SNP and gene transcription start site (TSS). |
7 |
af |
allele frequency of alternative allele (alt) |
8 |
pval_nominal |
p-value |
9 |
tissue_type |
tissue type |
1$ head gtex_v10_eqtl_hg38.txt
2chr position ref alt gene_name tss_distance af pval_nominal tissue_type
3chr1 766455 T C LINC01409 -12292 0.047058824 1.7230692640469627e-10 Vagina
4chr1 766938 C T LINC01409 -11809 0.047058824 7.331238896267609e-10 Vagina
5chr1 771358 T G LINC01409 -7389 0.047058824 3.298544072962652e-12 Vagina
6chr1 771398 G A LINC01409 -7349 0.67058825 2.133429762259741e-05 Vagina
7chr1 775571 G T LINC01409 -3176 0.047058824 3.298544072962652e-12 Vagina
8chr1 777550 T C LINC01409 -1197 0.05 9.539419071495843e-12 Vagina
9chr1 777751 A AT LINC01409 -996 0.05 9.539419071495843e-12 Vagina
10chr1 778534 A G LINC01409 -213 0.05 9.539419071495843e-12 Vagina
11chr1 778639 A G LINC01409 -108 0.08235294 2.6823764300049156e-08 Vagina
2.7.3.7.3 Risk SNP: txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
chr |
chromosome |
2 |
pos |
position |
3 |
rsId |
rsID identifier |
4 |
ref |
reference allele in the reference genome coordinate of the source cohort |
5 |
alt |
alternative allele in the reference genome coordinate of the source cohort. (This allele is the effect allele.) |
6 |
p |
p-value |
7 |
Trait |
trait |
8 |
Population |
population |
9 |
PMID |
PMID |
1$ head gwasatlas_v20191115_risk_snp_hg38.txt
2chr pos rsID ref alt p Trait Population PMID
3chr1 43718521 rs11420276 G GT 6.452e-13 Attention deficit hyperactivity disorder EUR 30478444
4chr1 96136884 rs1222063 A G 3.068e-08 Attention deficit hyperactivity disorder EUR 30478444
5chr3 20627579 rs4858241 G T 8.172e-09 Attention deficit hyperactivity disorder EUR 30478444
6chr4 31149834 rs28411770 C T 1.152e-08 Attention deficit hyperactivity disorder EUR 30478444
7chr5 88558577 rs4916723 A C 1.807e-08 Attention deficit hyperactivity disorder EUR 30478444
8chr5 88919777 rs304132 A G 3.047e-08 Attention deficit hyperactivity disorder EUR 30478444
9chr7 114418676 rs34291892 C CA 1.585e-08 Attention deficit hyperactivity disorder EUR 30478444
10chr8 34495092 rs74760947 A G 1.393e-08 Attention deficit hyperactivity disorder EUR 30478444
11chr10 104987596 rs11591402 A T 1.76e-08 Attention deficit hyperactivity disorder EUR 30478444
2.7.3.7.4 Enhancer (SEA v3): txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
chr |
chromosome |
2 |
start |
start position of enhancer |
3 |
end |
end position of enhancer |
4 |
associated_gene |
reference allele in the reference genome coordinate of the source cohort |
5 |
cell_tissue_type |
cell type/tissue type |
6 |
recognition_factor |
recognition factor (eg. h3k27ac) |
7 |
sequence_region |
sequence region (coding or noncoding) |
8 |
se_id |
SE ID of SEA |
1$ head sea_v3_enhancer_hg38.txt
2chr start end associated_gene cell_tissue_type recognition_factor sequence_region se_id
3chr10 88384139 88389120 RNLS 22Rv1 h3k27ac coding 442
4chr13 20117533 20129315 LINC01072 22Rv1 h3k27ac noncoding 443
5chr11 9056277 9061918 SCUBE2 22Rv1 h3k27ac coding 444
6chr5 44537047 44541439 LINC02224 22Rv1 h3k27ac noncoding 445
7chr9 112327808 112339994 PTBP3 22Rv1 h3k27ac coding 446
8chr4 138896634 138913955 LOC105377448 22Rv1 h3k27ac noncoding 447
9chr2 180254341 180260431 CWC22 22Rv1 h3k27ac coding 448
10chrX 66898375 66921461 EDA2R 22Rv1 h3k27ac coding 449
11chr7 12709011 12717389 ARL4A 22Rv1 h3k27ac coding 450
2.7.3.7.5 Enhancer (SEdb v2): txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
chr |
chromosome |
2 |
start |
start position of enhancer |
3 |
end |
end position of enhancer |
4 |
sample_id |
sample ID of SEdb |
5 |
se_id |
SE ID of SEdb |
6 |
cell_source |
source |
7 |
cell_type |
cell type |
8 |
tissue_type |
tissue type |
9 |
cell_state |
cell state |
1$ head sedb_v2_enhancer_hg38.txt
2chr start end sample_id se_id cell_source cell_type tissue_type cell_state
3chr6 32968553 32969528 SE_00_0001 TE_00_000100001 Roadmap Tissue Adipose adipose-tissue
4chr19 3404076 3405134 SE_00_0001 TE_00_000100002 Roadmap Tissue Adipose adipose-tissue
5chr22 17638273 17639305 SE_00_0001 TE_00_000100003 Roadmap Tissue Adipose adipose-tissue
6chr7 100428402 100429667 SE_00_0001 TE_00_000100004 Roadmap Tissue Adipose adipose-tissue
7chr19 6273122 6274837 SE_00_0001 TE_00_000100005 Roadmap Tissue Adipose adipose-tissue
8chr17 77128730 77140351 SE_00_0001 TE_00_000100006 Roadmap Tissue Adipose adipose-tissue
9chr6 33313122 33314294 SE_00_0001 TE_00_000100007 Roadmap Tissue Adipose adipose-tissue
10chr7 5555574 5556788 SE_00_0001 TE_00_000100008 Roadmap Tissue Adipose adipose-tissue
11chr7 143380426 143381762 SE_00_0001 TE_00_000100009 Roadmap Tissue Adipose adipose-tissue
2.7.3.7.6 Super enhancer (dbSUPER): txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
chr |
chromosome |
2 |
start |
start position of enhancer |
3 |
end |
end position of enhancer |
4 |
se_id |
SE ID of SEdb |
5 |
cell_type_type |
cell type/tissue type |
1$ head dbsuper_super_enhancer_hg38.txt
2chr start end se_id cell_type_type
3chr6 32580146 32643038 SE_10156 CD19 Primary
4chr14 105557581 105606092 SE_10157 CD19 Primary
5chr14 105677864 105749363 SE_10158 CD19 Primary
6chr6 167078442 167154502 SE_10159 CD19 Primary
7chr21 44137096 44181452 SE_10160 CD19 Primary
8chr5 150398244 150436858 SE_10161 CD19 Primary
9chr2 88831594 88886476 SE_10162 CD19 Primary
10chr6 33006818 33032650 SE_10163 CD19 Primary
11chr2 136114080 136141217 SE_10164 CD19 Primary
2.7.3.7.7 Super enhancer (SEA v3): txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
chr |
chromosome |
2 |
start |
start position of enhancer |
3 |
end |
end position of enhancer |
4 |
associated_gene |
reference allele in the reference genome coordinate of the source cohort |
5 |
cell_tissue_type |
cell type/tissue type |
6 |
recognition_factor |
recognition factor (eg. h3k27ac) |
7 |
sequence_region |
sequence region (coding or noncoding) |
8 |
se_id |
SE ID |
1$ head sea_v3_super_enhancer_hg38.txt
2chr start end associated_gene cell_tissue_type recognition_factor sequence_region se_id
3chr6 110617715 110700931 CDK19 22Rv1 h3k27ac coding 1
4chr7 92030110 92091121 AKAP9 22Rv1 h3k27ac coding 2
5chr11 59005426 59074536 LOC283194 22Rv1 h3k27ac noncoding 3
6chr5 71599725 71707973 MCCC2 22Rv1 h3k27ac coding 4
7chr21 6360657 6375827 CBS 22Rv1 h3k27ac coding 5
8chr12 101602935 101625047 MYBPC1 22Rv1 h3k27ac coding 6
9chr10 37145277 37199659 ANKRD30A 22Rv1 h3k27ac coding 7
10chr6 138221168 138289554 ARFGEF3 22Rv1 h3k27ac coding 8
11chr16 52550656 52582081 CASC16 22Rv1 h3k27ac noncoding 9
2.7.3.7.8 Super enhancer (SEdb v2): txt file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
chr |
chromosome |
2 |
start |
start position of enhancer |
3 |
end |
end position of enhancer |
4 |
sample_id |
sample ID of SEdb |
5 |
se_id |
SE ID of SEdb |
6 |
cell_source |
source |
7 |
cell_type |
cell type |
8 |
tissue_type |
tissue type |
9 |
cell_state |
cell state |
1$ head sedb_v2_super_enhancer_hg38.txt
2chr start end sample_id se_id cell_source cell_type tissue_type cell_state
3chr1 100008001 100081709 SE_02_1036 SE_02_103600569 NCBI GEO/SRA Cell line Mammary gland HCC70_XY018
4chr1 100015493 100079709 SE_02_1429 SE_02_142900169 NCBI GEO/SRA Cell line Blood GM12878_WT
5chr1 1000160 1006599 SE_02_0988 SE_02_098800774 NCBI GEO/SRA Cell line Blood K562_EPZ
6chr1 1000180 1006408 SE_02_1080 SE_02_108000734 NCBI GEO/SRA Cell line Muscle JR1 shCtrl
7chr1 100026929 100040607 SE_00_0009 SE_00_000900816 Roadmap Primary cell Blood CD8-positive-alpha-beta-T-cell
8chr1 100027783 100040448 SE_00_0027 SE_00_002700801 Roadmap Primary cell Blood natural-killer-cell
9chr1 100028493 100040305 SE_02_0707 SE_02_070700751 NCBI GEO/SRA Cell line Pancreas BxPC3 WT
10chr1 100028934 100040097 SE_02_0022 SE_02_002200606 NCBI GEO/SRA Primary cell Blood CD8donorA
11chr1 100033978 100061969 SE_02_1468 SE_02_146800857 NCBI GEO/SRA Cell line Blood HUDEP-2_WT
2.7.3.7.9 3D chromatin interaction: bed file (After decompression)
# |
Column name |
Description |
|---|---|---|
1 |
None |
chromosome (Interaction1) |
2 |
None |
start position of enhancer (Interaction1) |
3 |
None |
end position of enhancer (Interaction1) |
4 |
None |
chromosome (Interaction2) |
5 |
None |
start position of enhancer (Interaction2) |
6 |
None |
end position of enhancer (Interaction2) |
7 |
None |
Source/Interaction ID |
8 |
None |
Method |
9 |
None |
Tissue/cell type |
10 |
None |
Cell line |
1$ head 3D_hg19.bed
2chr1 37883731 37885731 chr1 38374488 38376488 3D_4DGenome_001 3C Kidney 293Trex
3chr1 68019395 68021395 chr1 68444820 68446820 3D_4DGenome_001 3C Kidney 293Trex
4chr1 94005332 94007332 chr1 94477646 94479646 3D_4DGenome_001 3C Kidney 293Trex
5chr1 9762548 9762685 chr1 9882283 9883893 3D_OncoBase_084 EpiTensor Kidney Kidney
6chr1 9848832 9851345 chr1 9882283 9883893 3D_OncoBase_084 EpiTensor Kidney Kidney
7chr1 98991643 98992662 chr1 99114108 99115246 3D_OncoBase_084 EpiTensor Kidney Kidney
8chr1 99114108 99115246 chr1 99125090 99125899 3D_OncoBase_084 EpiTensor Kidney Kidney
9chr1 98991643 98992662 chr1 99125090 99125899 3D_OncoBase_084 EpiTensor Kidney Kidney
10chr1 99181550 99181760 chr1 99182450 99183081 3D_OncoBase_084 EpiTensor Kidney Kidney
11chr1 99125090 99125899 chr1 99193746 99195271 3D_OncoBase_084 EpiTensor Kidney Kidney
2.7.3.7.10 MPRA: csv file
Download source: https://mpravardb.rc.ufl.edu/
# |
Column name |
Description |
|---|---|---|
1 |
chr |
chromosome |
2 |
pos |
position of variant |
3 |
ref |
reference allele in the reference genome coordinate of the source cohort |
4 |
alt |
alternative allele in the reference genome coordinate of the source cohort. (This allele is the effect allele.) |
5 |
genome |
reference genome |
6 |
rsid |
rsID identifier |
7 |
disease |
trait/disease |
8 |
cellline |
cell line |
9 |
Description |
description |
10 |
log2FC |
Log2(Fold change) |
11 |
pvalue |
P value |
12 |
fdr |
FDR |
13 |
MPRA_study |
MPRA study |
1$ head All_MPRA_Data.csv
2"chr","pos","ref","alt","genome","rsid","disease","cellline","Description","log2FC","pvalue","fdr","MPRA_study"
3"1",2440958,"A","G","hg38","rs6688934","Schizophrenia","SH-SY5Y","1,049 SZ and 30 AD variants in 64 SZ loci and 9 AD loci, respectively",NA,0.108571634,0.341634497,"A screen of 1049 schizophrenia and 30 Alzheimer's-associated variants for regulatory potential (Myint et al., 2020)"
4"1",2441515,"A","G","hg38","rs6673661","Schizophrenia","SH-SY5Y","1,049 SZ and 30 AD variants in 64 SZ loci and 9 AD loci, respectively",NA,0.057599896,0.234108669,"A screen of 1049 schizophrenia and 30 Alzheimer's-associated variants for regulatory potential (Myint et al., 2020)"
5"1",2443319,"A","G","hg38","rs4648844","Schizophrenia","SH-SY5Y","1,049 SZ and 30 AD variants in 64 SZ loci and 9 AD loci, respectively",NA,0.014320564,0.115533569,"A screen of 1049 schizophrenia and 30 Alzheimer's-associated variants for regulatory potential (Myint et al., 2020)"
6"1",2444405,"T","G","hg38","rs6687012","Schizophrenia","SH-SY5Y","1,049 SZ and 30 AD variants in 64 SZ loci and 9 AD loci, respectively",NA,0.258798019,0.530956548,"A screen of 1049 schizophrenia and 30 Alzheimer's-associated variants for regulatory potential (Myint et al., 2020)"
7"1",2448266,"A","G","hg38","rs942820","Schizophrenia","SH-SY5Y","1,049 SZ and 30 AD variants in 64 SZ loci and 9 AD loci, respectively",NA,0.077694104,0.275581292,"A screen of 1049 schizophrenia and 30 Alzheimer's-associated variants for regulatory potential (Myint et al., 2020)"
8"1",2455662,"C","T","hg38","rs4648845","Schizophrenia","SH-SY5Y","1,049 SZ and 30 AD variants in 64 SZ loci and 9 AD loci, respectively",NA,0.453624774,0.700344436,"A screen of 1049 schizophrenia and 30 Alzheimer's-associated variants for regulatory potential (Myint et al., 2020)"
9"1",8362616,"T","C","hg38","rs2252865","Schizophrenia","SH-SY5Y","1,049 SZ and 30 AD variants in 64 SZ loci and 9 AD loci, respectively",NA,0.551078425,0.775862448,"A screen of 1049 schizophrenia and 30 Alzheimer's-associated variants for regulatory potential (Myint et al., 2020)"
10"1",8363450,"A","G","hg38","rs10779702","Schizophrenia","SH-SY5Y","1,049 SZ and 30 AD variants in 64 SZ loci and 9 AD loci, respectively",NA,0.295545372,0.575535724,"A screen of 1049 schizophrenia and 30 Alzheimer's-associated variants for regulatory potential (Myint et al., 2020)"
11"1",8372076,"C","T","hg38","rs894875","Schizophrenia","SH-SY5Y","1,049 SZ and 30 AD variants in 64 SZ loci and 9 AD loci, respectively",NA,0.543395748,0.774441451,"A screen of 1049 schizophrenia and 30 Alzheimer's-associated variants for regulatory potential (Myint et al., 2020)"